Najmanovich Research Group
RESOURCES
FlexAID
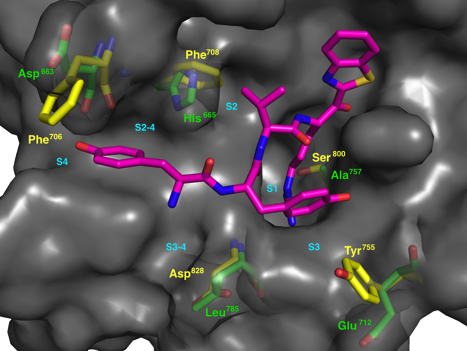
Small-molecule protein docking is an essential tool in drug design and to understand molecular recognition. Our group is responsible for the development of FlexAID [36]. FlexAID, is a docking algorithm that can use small-molecules and peptides as ligands and proteins/nucleic acids as targets. It permits full ligand flexibility as well as target side-chain flexibility. FlexAID utilizes a soft scoring function, i.e. one that is not highly dependent on specific geometric criteria, based on surface complementarity. The energy parameters of the scoring function were derived from the classification of a largedataset of native and near native (less than 2Å RMSD) conformations for nearly 1500 complexes from the PDBbind database as true positive examples. These were countered over successive rounds of Monte Carlo optimization over an ever increasing and successively more difficult sets (increasingly lower energy decoys) with RMSD above 2Å. FlexAID has been used in numerous projects in collaborations and in our group.
FlexAID has been shown to outperform existing widely used software such as AutoDock Vina and FlexX in the prediction of binding poses. This is particularly true in cases where target flexibility is crucial, such as is likely to be the case when using homology models. The FlexAID source code is available at our GitHub page.
NRGsuite
The NRGsuite is a PyMOL plugin that permits the detection of surface cavities in macromolecules using GetCleft, their refinement, calculation of volume and use, individually or jointly, as target binding-sites for docking simulations with FlexAID. The NRGsuite offers users control over a large number of important parameters in docking simulations including the assignment of flexile side-chains and the definition of geometric constraints (to simulate covalent docking). Furthermore, the NRGsuite permits the visualization of the docking simulation in real time.
The NRGsuite offers a convenient and user-friendly manner to perform docking simulations for structure-based drug design and the exploration of FlexAID simulation parameters as well as preparation of input files for large-scale runs of FlexAID on its own (for virtual screening). The NRGsuite is also an excellent tool for teaching. Please take the time to cite FlexAID and the NRGsuite [40] if you use it. You can download the user manual to get started and the executables with the buttons below.
GetCleft
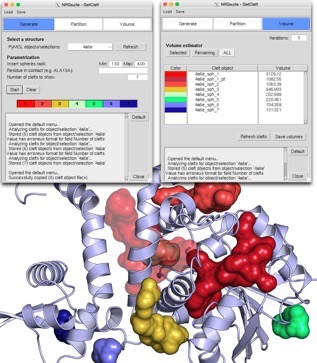
The detection of cavities, both internal or surface-exposed is very useful. Our GetCleft software is an implementation in C of the SurfNet algorithm developed by Laskowski [Laskowski, R. Surfnet - a Program for Visualizing Molecular-Surfaces, Cavities, and Intermolecular Interactions. 1995;13: 323–330]. We provide it here separately as it is a necessary independent step in the use of IsoMIF or FlexAID. However, if you use the NRGsuite, GetCleft is built-in and also gives user the possiblity to refine and measure the volume of cavities as shown in the image above. The GetCleft source code is available at our GitHub page.
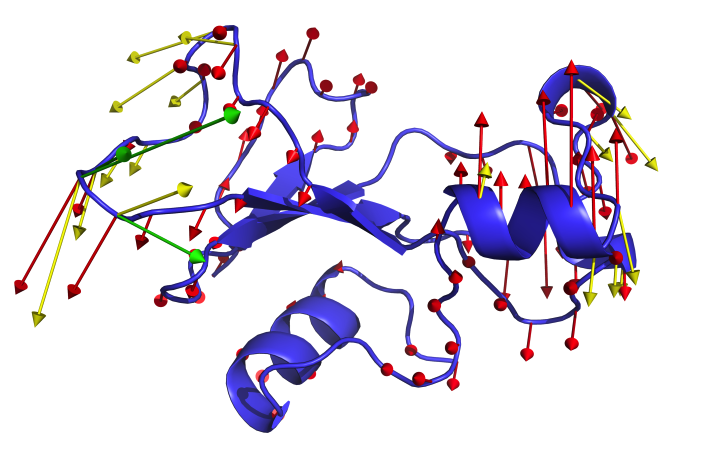
ENCoM
ENCoM is a coarse-grained normal mode analysis method that is unique in the sense that the elastic constants the define the modes of movements are modulated by a potential force field and the surface area in contact between atoms belonging to different residues in the protein or nucleotides in RNA. The method has been applied to a number of questions, primarily thus far to proteins. We have used ENCoM to predict the effect of mutations on stability and applied it as a tool in protein engineering. We also use it to generate conformational ensembles that may be useful for a number of applications, including in the representation of ensembles for virtual screening. We are currently applying ENCoM to study allostery as well as signal transduction in GPCRs. Whereas we have developed a very easy to use and convenient web-service to predict the effect of mutations and generate conformational ensembles, this is currently temporarily unavailable. All the results that could be obtained through the web interface can also be obtained via the stand-alone version of ENCoM that can be downloaded from GitHub (includes the necessary scripts). In order to facilitate its use, we have created a step-by-step tutorial. If you use ENCoM, please cite the original article [31] as well as the tutorial article [42] for which you will the scripts here.
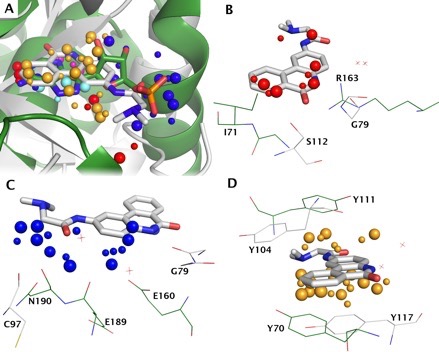
IsoMIF
IsoMIF identifies binding site molecular interaction field similarities between proteins. IsoMIF allows you to identify binding site molecular interaction field (MIF) similarities between pairs of structures. You can also run your query structure against a database of pre-calculated MIFs or all-against-all comparisons of you own custom PDB entries. As an example of IsoMIF, the figure shows the superimposition of the 3GEY and 1AHB (panel A) based on the MIF similarities found for 5 probes. The H-bond acceptor probes matched (Pannel B) shows the H-bonds made by 3GEY with the backbone amide of glycine 79 could be made by arginine 163 of 1AHB. While it doesn’t seem to be used in binding Pj34, the donor hydroxyl of serine 112 seems to have a counterpart with the backbone amide of isoleucine 71. Similarly there are many H-bond donor probes found and Figure (panel C) shows the acceptors atoms in each structure that are responsible for these similarities. Shared aromatic interactions are the result of favourable interaction energies between aromatic probes and tyrosine 117 or tyrosine 70 respectively that can make face-to-face aromatic interactions with the inhibitor (panel D). We deeply regret that the IsoMIF Server is not currently available due to technical problems. In the meantime, you can download the software to run locally and also the datasets of pre-calculated MIFs to run your query against. The,source code and scripts to visualize MIFs as well as matched similarities (release 150311) can be downloaded from GitHub. You can find here the Instructions to compile and run IsoMIF locally. The following datasets used in the article above to compare IsoMIF to other methods can be downloaded here containing: 1. Entries in the Kahraman, Homogenous, Steroid, Soippa, PDBbind and scPDB datasets (PDB, residue name, residue number, chain, alternate location); 2. True positive pairs for scPDB dataset; and 3. True positive pairs for PDBbind dataset.
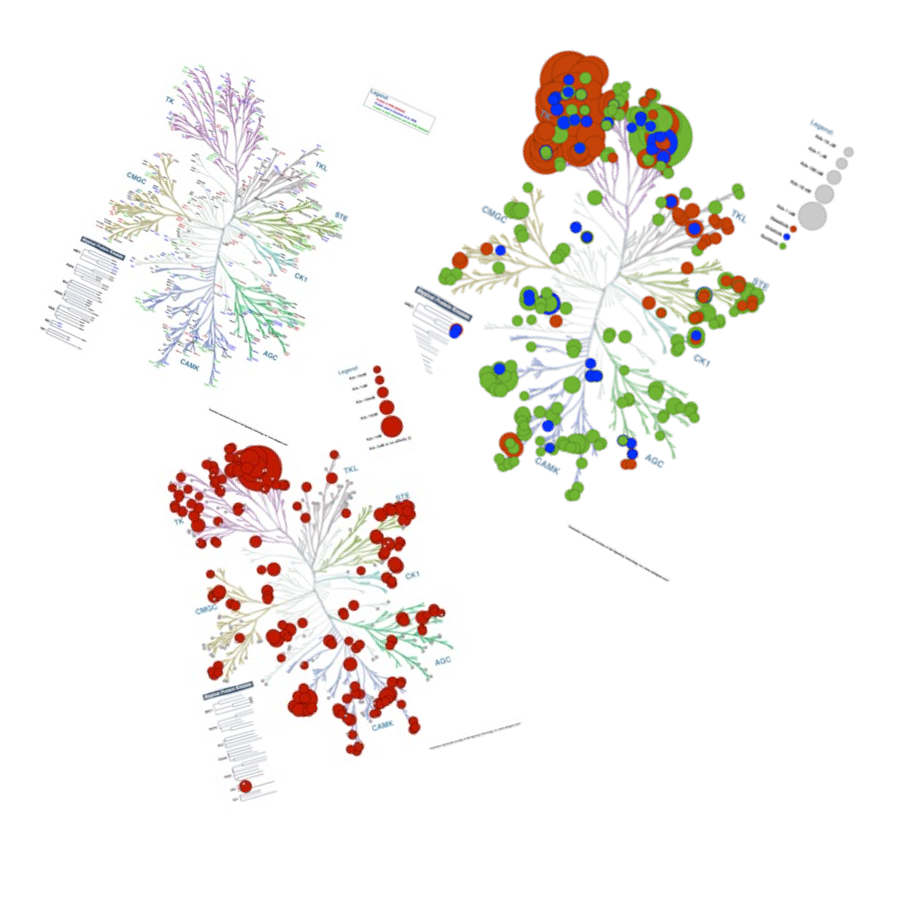
Kinome Render
Human protein kinases play fundamental roles mediating the majority of signal transduction pathways in eukaryotic cells as well as a multitude of other processes involved in metabolism, cell-cycle regulation, cellular shape, motility, differentiation and apoptosis. The human protein kinome contains 518 members. Most studies that focus on the human kinome require, at some point, the visualization of large amounts of data. The visualization of such data within the framework of a phylogenetic tree may help identify key relationships between different protein kinases in view of their evolutionary distance and the information used to annotate the kinome tree. For example, studies that focus on the promiscuity of kinase inhibitors can benefit from the annotations to depict binding affinities across kinase groups. Images involving the mapping of information into the kinome tree are common. However, producing such figures manually can be a long arduous process prone to errors. To circumvent this issue, we have developed a stand-alone tool called Kinome Render (KR) that produces customized annotations on the human kinome tree. KR allows the creation and automatic overlay of customizable text or shape-based annotations of different sizes and colors on the human kinome tree.